Хроматография (от греч. chroma, родительный падеж chromatos — цвет, краска и ...графия), физико-химический метод разделения и анализа смесей, основанный на распределении их компонентов между двумя фазами — неподвижной и подвижной (элюент), протекающей через неподвижную.
Историческая справка. Метод разработан в 1903 М. Цветом, который показал, что при пропускании смеси растительных пигментов через слой бесцветного сорбента индивидуальные вещества располагаются в виде отдельных окрашенных зон. Полученный таким образом послойно окрашенный столбик сорбента Цвет назвал хроматограммой, а метод — Х. Впоследствии термин «хроматограмма» стали относить к разным способам фиксации результатов многих видов Х. Однако вплоть до 40-х гг. Х. не получила должного развития. Лишь в 1941 А. Мартин и Р. Синг открыли метод распределительной Х. и показали его широкие возможности для исследования белков и углеводов. В 50-е гг. Мартин и американский учёный А. Джеймс разработали метод газо-жидкостной Х.
Основные виды Х. В зависимости от природы взаимодействия, обусловливающего распределение компонентов между элюентом и неподвижной фазой, различают следующие основные виды Х. — адсорбционную, распределительную, ионообменную, эксклюзионную (молекулярно-ситовую) и осадочную. Адсорбционная Х. основана на различии сорбируемости разделяемых веществ адсорбентом (твёрдое тело с развитой поверхностью); распределительная Х. — на разной растворимости компонентов смеси в неподвижной фазе (высококипящая жидкость, нанесённая на твёрдый макропористый носитель) и элюенте (следует иметь в виду, что при распределительном механизме разделения на перемещение зон компонентов частичное влияние оказывает и адсорбционное взаимодействие анализируемых компонентов с твёрдым сорбентом); ионообменная Х. — на различии констант ионообменного равновесия между неподвижной фазой (ионитом) и компонентами разделяемой смеси; эксклюзионная (молекулярно-ситовая) Х. — на разной проницаемости молекул компонентов в неподвижную фазу (высокопористый неионогенный гель). Эксклюзионная Х. подразделяется на гель-проникающую (ГПХ), в которой элюент — неводный растворитель, и гель-фильтрацию, где элюент — вода. Осадочная Х, основана на различной способности разделяемых компонентов выпадать в осадок на твёрдой неподвижной фазе.
В соответствии с агрегатным состоянием элюента различают газовую и жидкостную Х. В зависимости от агрегатного состояния неподвижной фазы газовая Х. бывает газо-адсорбционной (неподвижная фаза — твёрдый адсорбент) и газожидкостной (неподвижная фаза — жидкость), а жидкостная Х. — жидкостно-адсорбционной (или твёрдо-жидкостной) и жидкостно-жидкостной. Последняя, как и газо-жидкостная, является распределительной Х. К твёрдо-жидкостной Х. относятся тонкослойная и бумажная.
Различают колоночную и плоскостную Х. В колоночной сорбентом заполняют специальные трубки — колонки, а подвижная фаза движется внутри колонки благодаря перепаду давления. Разновидность колоночной Х. — капиллярная, когда тонкий слой сорбента наносится на внутренние стенки капиллярной трубки. Плоскостная Х. подразделяется на тонкослойную и бумажную. В тонкослойной Х. тонкий слой гранулированного сорбента или пористая плёнка наносится на стеклянную или металлическую пластинки; в случае бумажной Х. используют специальную хроматографическую бумагу. В плоскостной Х. перемещение подвижной фазы происходит благодаря капиллярным силам.
При хроматографировании возможно изменение по заданной программе температуры, состава элюента, скорости его протекания и др. параметров.
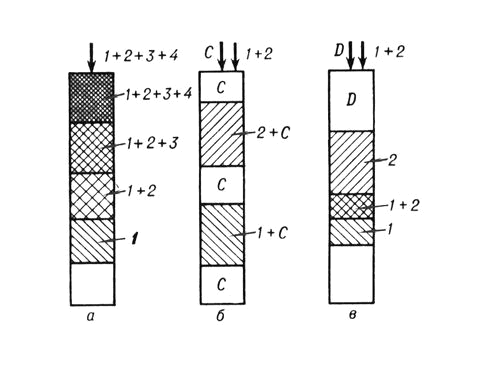
В зависимости от способа перемещения разделяемой смеси вдоль слоя сорбента различают следующие варианты Х.: фронтальный, проявительный и вытеснительный. При фронтальном варианте в слой сорбента непрерывно вводится разделяемая смесь, состоящая из газа-носителя и разделяемых компонентов, например 1, 2, 3, 4, которая сама является подвижной фазой. Через некоторое время после начала процесса наименее сорбируемый компонент (например, 1) опережает остальные и выходит в виде зоны чистого вещества раньше всех, а за ним в порядке сорбируемости последовательно располагаются зоны смесей компонентов: 1 + 2, 1 + 2 + 3, 1 + 2 + 3 + 4 (рис., a). При проявительном варианте через слой сорбента непрерывно проходит поток элюента и периодически в слой сорбента вводится разделяемая смесь веществ. Через определённое время происходит деление исходной смеси на чистые вещества, располагающиеся отдельными зонами на сорбенте, между которыми находятся зоны элюента (рис., б). При вытеснительном варианте в сорбент вводится разделяемая смесь, а затем поток газа-носителя, содержащего вытеснитель (элюент), при движении которого смесь через некоторый период времени разделится на зоны чистых веществ, между которыми окажутся зоны их смеси (рис., в). Ряд видов Х. осуществляется с помощью приборов, называемых хроматографами, в большинстве из которых реализуется проявительный вариант Х. Хроматографы используют для анализа и для препаративного (в т. ч. промышленного) разделения смесей веществ. При анализе разделённые в колонке хроматографа вещества вместе с элюентом попадают через различные промежутки времени в установленное на выходе из хроматографической колонки детектирующее устройство, регистрирующее их концентрации во времени. Полученную в результате этого выходную кривую называют хроматограммой. Для качественного хроматографического анализа определяют время от момента ввода пробы до выхода каждого компонента из колонки при данной температуре и при использовании определённого элюента. Для количественного анализа определяют высоты или площади хроматографических пиков с учётом коэффициентов чувствительности используемого детектирующего устройства к анализируемым веществам.
Для анализа и разделения веществ, переходящих без разложения в парообразное состояние, наибольшее применение получила газовая Х., где в качестве элюента (газа-носителя) используются гелий, азот, аргон и др. газы. Для газо-адсорбционного варианта Х. в качестве сорбента (частицы диаметром 0,1—0,5 мм) используют силикагели, алюмогели, молекулярные сита, пористые полимеры и др. сорбенты с удельной поверхностью 5—500 м2/г. Для газо-жидкостной Х. сорбент готовят нанесением жидкости в виде плёнки (высококипящие углеводороды, сложные эфиры, силоксаны и др.) толщиной несколько мкм на твёрдый носитель с удельной поверхностью 0,5—5 м2/г и более. Рабочие температурные пределы для газо-адсорбционного варианта Х. от —70 до 600 °С, для газо-жидкостного от —20 до 400 °С. Газовой Х. можно разделить несколько см3 газа или мг жидких (твёрдых) веществ; время анализа от несколькихсек до нескольких часов.
В жидкостной колоночной Х. в качестве элюента применяют легколетучие растворители (например, углеводороды, эфиры, спирты), а в качестве неподвижной фазы — силикагели (в т. ч. силикагели с химически привитыми к поверхности различными функциональными группами — эфирными, спиртовыми и др.), алюмогели, пористые стекла; размер частиц всех этих сорбентов несколько мкм. Подавая элюент под давлением до 50 Мн/м2 (500 кгс/см2), удаётся сократить время анализа от 2—3 ч до нескольких мин. Для повышения эффективности разделения сложных смесей используют программируемое во времени изменение свойств элюента путём смешения растворителей разной полярности (градиентное элюирование).
Жидкостная молекулярно-ситовая Х. отличается использованием сорбентов, имеющих поры строго определённого размера (пористые стекла, молекулярные сита, в том числе декстрановые и др. гели). В тонкослойной и бумажной Х. исследуемую смесь в жидком виде наносят на стартовую линию (начало пластинки или полоски бумаги), а затем разделяют на компоненты восходящим или нисходящим потоком элюента. Последующее обнаружение (проявление) разделённых веществ на хроматограмме (так в этих случаях называют пластину с нанесённым на неё сорбентом или хроматографическую бумагу, на которых произошло разделение исследуемой смеси на компоненты) осуществляют при помощи ультрафиолетовой (УФ) спектроскопии, инфракрасной (ИК) спектроскопии или обработкой реактивами, образующими с анализируемыми веществами окрашенные соединения.
Качественно состав смесей с помощью этих видов Х. характеризуют определённой скоростью перемещения пятен веществ относительно скорости движения растворителя в данных условиях. Количественный анализ осуществляют измерением интенсивности окраски вещества на хроматограмме.
Х. широко применяется в лабораториях и в промышленности для качественного и количественного анализа многокомпонентных систем, контроля производства, особенно в связи с автоматизацией многих процессов, а также для препаративного (в т. ч. промышленного) выделения индивидуальных веществ (например, благородных металлов), разделения редких и рассеянных элементов.
Газовая Х. применяется для газов разделения, определения примесей вредных веществ в воздухе, воде, почве, промышленных продуктах; определения состава продуктов основного органического и нефтехимического синтеза, выхлопных газов, лекарственных препаратов, а также в криминалистике и т.д. Разработаны аппаратура и методики анализа газов в космических кораблях, анализа атмосферы Марса, идентификации органических веществ в лунных породах и т.п.
Газовая Х. применяется также для определения физико-химических характеристик индивидуальных соединений: теплоты адсорбции и растворения, энтальпии, энтропии, констант равновесия и комплексообразования; для твёрдых веществ этот метод позволяет измерить удельную поверхность, пористость, каталитическую активность.
Жидкостная Х. используется для анализа, разделения и очистки синтетических полимеров, лекарственных препаратов, детергентов, белков, гормонов и др. биологически важных соединений. Использование высокочувствительных детекторов позволяет работать с очень малыми количествами веществ (10-11—10-9 г), что исключительно важно в биологических исследованиях. Часто применяется молекулярно-ситовая Х. и Х. по сродству; последняя основана на способности молекул биологических веществ избирательно связываться друг с другом.
Тонкослойная и бумажная Х. используются для анализа жиров, углеводов, белков и др. природных веществ и неорганических соединений.
В некоторых случаях для идентификации веществ используется Х. в сочетании с др. физико-химическими и физическими методами, например с масс-спектрометрией, ИК-, УФ-спектроскопией и др. Для расшифровки хроматограмм и выбора условий опыта применяют ЭВМ.
Лит.: Жуховицкий А. А., Туркельтауб Н. М., Газовая хроматография, М., 1962; Киселев А. В., Яшин Я. И., Газо-адсорбционная хроматография, М., 1967; Сакодынский К. И., Волков С. А., Препаративная газовая хроматография, М., 1972; Гольберт К. А., Вигдергауз М. С., Курс газовой хроматографии, М., 1974; Хроматография на бумаге, пер. с чеш., М., 1962; Детерман Г., Гель-хроматография, пер. с нем., М., 1970; Morris С. J. О., Morris P., Separation methods in biochemistry, L., 1964.
К. И. Сакодынский.
Основные варианты проведения хроматографического процесса: а — фронтальный; б — проявительный; в — вытеснительный; 1, 2, 3, 4 — разделяемые вещества; C — несорбирующаяся подвижная фаза; D — вытеснитель.
Хроматографы, приборы или установки для хроматографического разделения и анализа смесей веществ (см. Хроматография). Основными частями Х. являются: система для ввода исследуемой смеси веществ (пробы); хроматографическая колонка; детектирующее устройство (детектор); системы регистрации и термостатирования; для препаративных (в т. ч. производственных) Х., кроме того, отборные приспособления и приёмники для разделённых компонентов.
В соответствии с агрегатным состоянием используемой подвижной фазы существуют газовые и жидкостные Х. В подавляющем числе Х. реализуется проявительный вариант хроматографии. В газовом Х. (см. рис.) газ-носитель из баллона через регуляторы расхода и давления непрерывно с постоянной или переменной скоростью подаётся в хроматографическую колонку-трубку (диаметром 2—5 мм и длина 1—10 м), заполненную сорбентом и помещенную в термостат, позволяющий поддерживать заданную температуру (вплоть до 500 °С).
Ввод газообразной пробы (1—50 см3) и жидкой (несколько мкл) осуществляется либо вручную (газовым шприцем или микрошприцем), либо автоматически — при помощи микродозаторов. В хроматографической колонке происходит разделение исходной многокомпонентной смеси на ряд бинарных смесей, состоящих из газа-носителя и одного из анализируемых компонентов. Бинарные смеси в определённой последовательности, зависящей от сорбируемости компонентов, поступают в детектор. В результате происходящих в детекторе процессов (изменения теплопроводности, ионизационного тока и др.) фиксируется изменение концентрации выходящих компонентов; преобразованные в электрический сигнал, эти процессы записываются в виде выходной кривой.
Наиболее распространённые детекторы газовых Х. — термокондуктометрические и ионизационные. Типичным примером первых является детектор по теплопроводности (катарометр), в мостовую цепь которого включены две ячейки для измерения теплопроводности; через них протекают потоки чистого газа-носителя и бинарная смесь. Теплопроводность последней отличается от теплопроводности чистого газа-носителя; поэтому при прохождении бинарной смеси через чувствительный элемент детектора — нагретую спираль с сопротивлением 10—80 ом — меняются температура и сопротивление спирали в зависимости от концентрации компонента. Такой детектор позволяет определять концентрации веществ в пределах 10-1—10-2%.
Главной частью ионизационных детекторов является ионизационная камера, где происходит ионизация молекул, попадающих в неё с потоком газа-носителя из хроматографической колонки. Ионизацию исследуемых веществ осуществляют в пламени водорода, метастабильными атомами аргона или гелия, медленными электронами и т.д. Ионы под воздействием приложенного напряжения перемещаются в ионизационной камере, что приводит к образованию электрического тока. Ионизационные детекторы позволяют определять концентрации веществ в пределах 10-4—10-7%.
Термокондуктометрические и ионизационные детекторы характеризуются чувствительностью (минимально определяемая концентрация вещества), селективностью (способность избирательно определять в смеси отдельные компоненты), прямой зависимостью сигнала от концентрации.
В жидкостном Х. в качестве детектирующего устройства используют проточный рефрактометр, включаемый по дифференциальной схеме, или детектор поглощения в ультрафиолетовой области. Подачу подвижной фазы — растворителя осуществляют при помощи беспульсационных систем (давление до 50 Мн/м2, или 500 кгс/см2), а ввод пробы — микрошприцем или переключающимся краном. Длина хроматографической колонки в жидкостном Х. не превышает 1 м. В целом детекторы жидкостных Х. обладают существенно меньшей чувствительностью (примерно на 2 порядка), чем детекторы газовых Х. Для точного измерения концентраций веществ детекторы калибруют по смесям известного состава.
Достигаемые скорость и точность анализа в Х. во многом определяются правильным выбором рабочего режима детектора и условий эксперимента (тип сорбента, температура, скорость газа-носителя, длина хроматографической колонки и др.). Для ускорения анализа применяют программированное во времени изменение температуры хроматографической колонки или расхода газа-носителя.
Лит.: Приборы для хроматографии, М., 1973; Бражников В. В., Дифференциальные детекторы для газовой хроматографии, М., 1974.
К. И. Сакодынский.
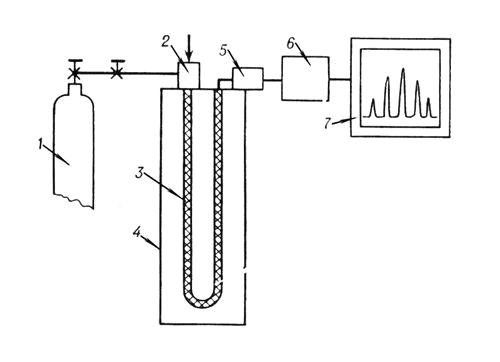
Принципиальная схема газового хроматографа: 1 — баллон с инертным газом; 2 — устройство для ввода пробы в хромотографическую колонку; 3 — хромотографическая колонка; 4 — термостат; 5 — детектор; 6 — преобразователь сигналов; 7 — регистратор.
Адсорбенты, искусственные и природные тела с развитой поверхностью, которая хорошо поглощает (адсорбирует) вещества из газов и растворов, окружающих А. Адсорбционные свойства А. зависят от химического состава и физического состояния поверхности, от характера пористости и удельной поверхности (поверхности, приходящейся на 1 г вещества). Непористые А. (молотые кристаллы, мелкокристаллические осадки, частицы дымов, сажи, аэросил) имеют удельные поверхности приблизительно от 1 м2/г до 500 м2/г. Удельная поверхность пористых А. (силикагелей, алюмогелей, алюмо-силикатных катализаторов, активных углей) достигает 1000 м2/г. Непористые высокодисперсные А. получают главным образом при термическом разложении или неполном сгорании углеводородов (получение саж), при сжигании элементоорганических или галогенных соединений (получение высокодисперсного кремнезёма — аэросила). Пористые А. получают следующими способами: 1) создавая сети пор в грубодисперсных твёрдых телах химическим воздействием (см. Активный уголь); 2) приготавливая гели из коллоидных растворов — золей; при высушивании таких гелей из зазоров между коллоидными частицами удаляется растворитель и вследствие этого получаемый материал обладает развитой системой пор; 3) синтезируя пористые кристаллы типа цеолитов, приобретающие особенно большое значение как катализаторы, А. и молекулярные сита. А. получают также термическим разложением карбонатов, оксалатов, гидроокисей, некоторых полимеров, молекулярной возгонкой твёрдых тел в вакууме и другими способами.
А. применяют как носители в катализе, как наполнители для полимеров, для хроматографического разделения смесей (см. Хроматография), в противогазах, в медицине (см. Адсорбирующие средства в медицине), в нефтехимии для очистки нефтепродуктов и газов, а также в высоковакуумной технике для сорбционных насосов.
В.И. Шимулис.
Декстран, (C6H10O5)n, полисахарид бактериального происхождения, полимер глюкозы. Молярная масса достигает 10 млн. В линейной части молекулы Д. остатки глюкозы соединены связями между 1 и 6-м атомами углерода; ветвление обусловлено связями между 1 и 4-м, 1 и 3-м, 1 и 2-м атомами. Д. получают при культивировании на искусственной питательной среде микроорганизма рода Leuconostoc. В виде частично гидролизованного раствора с молярной массой около 60 тыс. и количеством 1, 6-связей не менее 90% Д. применяется в клинике как заменитель плазмы крови. Препарат обеспечивает нормальное осмотическое давление, соответствующее осмотическому давлению крови. В хроматографии используются модифицированные Д. — так называемые сефадексы (см. Молекулярные сита).
Количественный анализ, совокупность химических, физико-химических и физических методов определения количественного соотношения компонентов, входящих в состав анализируемого вещества. Наряду с качественным анализом К. а. является одним из основных разделов аналитической химии. По количеству вещества, взятого для анализа, различают макро-, полумикро-, микро- и ульт-рамикрометоды К. а. В макрометодах масса пробы составляет обычно >100 мг, объём раствора > 10 мл; в ультрамикрометодах — соответственно 1—10-1 мг и 10-3—10-6 мл (см. также Микрохимический анализ, Ультрамикрохимический анализ). В зависимости от объекта исследования различают неорганический и органический К. а., разделяемый, в свою очередь, на элементный, функциональный н молекулярный анализ. Элементный анализ позволяет установить содержание элементов (ионов), функциональный анализ — содержание функциональных (реакционноспособных) атомов и групп в анализируемом объекте. Молекулярный К. а. предусматривает анализ индивидуальных химических соединений, характеризующихся определенной молекулярной массой. Важное значение имеет так называемый фазовый анализ — совокупность методов разделения и анализа отдельных структурных (фазовых) составляющих гетерогенных систем. Помимо специфичности и чувствительности (см. Качественный анализ), важная характеристика методов К. а. — точность, то есть значение относительной ошибки определения; точность и чувствительность в К. а. выражают в процентах.
К классическим химическим методам К. а. относятся: гравиметрический анализ, основанный на точном измерении массы определяемого вещества, и объёмный анализ. Последний включает титриметрический объёмный анализ — методы измерения объёма раствора реагента, израсходованного на реакцию с анализируемым веществом, и газовый объёмный анализ — методы измерения объёма анализируемых газообразных продуктов (см. Титриметрический анализ, Газовый анализ).
Наряду с классическими химическими методами широко распространены физические и физико-химические (инструментальные) методы К. а., основанные на измерении оптических, электрических, адсорбционных, каталитических и других характеристик анализируемых веществ, зависящих от их количества (концентрации). Обычно эти методы делят на следующие группы: электрохимические (кондуктометрия, полярография, потенциометрия и др.); спектральные или оптические (эмиссионный и абсорбционный спектральный анализ, фотометрия, колориметрия, нефелометрия, люминесцентный анализ и др.); рентгеновские (абсорбционный и эмиссионный рентгеноспектральный анализ, рентгенофазовый анализ и др.); хроматографический (жидкостная, газовая, газо-жидкостная хроматография и др.); радиометрические (активационный анализ и др.); масс-спектрометрические. Перечисленные методы, уступая химическим в точности, существенно превосходят их по чувствительности, избирательности, скорости выполнения. Точность химических методов К. а. находится обычно в пределах 0,005—0,1%; ошибки определения инструментальными методами составляют 5—10%, а иногда и значительно больше. Чувствительность некоторых методов К. а. приведена ниже (%):
Объёмный.......................................................10-1
Гравиметрический......................................... 10-2
Эмиссионный спектральный.........................10-4
Абсорбционный рентгеноспектральный...... 10-4
Масс-спектрометрический.............................10-4
Кулонометрический....................................... 10-5
Люминесцентный.......................................... 10-6—10-5
Фотометрический колориметрический......... 10-7—10-4
Полярографический.........................................10-8—10-6
Активационный................................................10-9—10-8
При использовании физических и физико-химических методов К. а. требуются, как правило, микроколичества веществ. Анализ может быть в ряде случаев выполнен без разрушения пробы; иногда возможна также непрерывная и автоматическая регистрация результатов. Эти методы используются для анализа веществ высокой чистоты, оценки выходов продукции, изучения свойств и строения веществ и т.д.
В. В. Краснощёков.
Молекулярные сита, сорбенты, избирательно поглощающие из окружающей среды вещества, молекулы которых не превышают определённых размеров. Такие сорбенты как бы отсеивают крупные молекулы от мелких. Различают минеральные (неорганические) и органические М. с. Неорганические М. с. имеют жёсткую кристаллическую структуру, в которой находятся полости, соединённые между собой узкими каналами «порами» или «окнами». Малые размеры «окон» препятствуют диффузии крупных молекул во внутренние полости сорбента. Некоторые алюмосиликаты — природные и синтетические цеолиты — характерные представители М. с. этого типа.
Органические М. с. — гелевидные сорбенты, получаемые на основе высокомолекулярных соединений. Структура таких сорбентов представляет собой пространственную сетку из цепочечных макромолекул, «сшитых» в отдельных точках химическими связями. Из гелевидных М. с. промышленного производства наиболее распространены различные типы сефадекса — сорбента на основе декстрана (высокомолекулярного полисахарида). М. с., содержащие ионогенные (диссоциирующие на ионы) группы и способные к ионному обмену, называют ионитовыми ситами. В отличие от обычных ионитов, они избирательно поглощают из раствора лишь достаточно малые ионы, исключая из ионообменного процесса крупные ионы, диффузия которых сквозь структурную сетку сорбента затруднена.
М. с. выпускают в виде порошка, зёрен неправильной формы, сферических гранул. Их используют для очистки веществ от нежелательных примесей, фракционирования синтетических полимеров, хроматографического разделения белков, углеводов, гормонов, антибиотиков и пр.
Лит.: Детерман Г., Гель-хроматография, пер. с нем., М., 1970.
Л. А. Шиц.
Поверхность удельная, усреднённая характеристика размеров внутренних полостей (каналов, пор) пористого тела или частиц раздробленной фазы дисперсной системы. П. у. выражают отношением общей поверхности пористого или диспергированного в данной среде тела к его объёму или массе. П. у. пропорциональна дисперсности или, что то же, обратно пропорциональна размеру частиц дисперсной фазы. От величины П. у. зависят поглотительная способность адсорбентов, эффективность твёрдых катализаторов, свойства фильтрующих материалов. П. у. активных углей составляет 500—1500, силикагелей — до 800, макропористых ионообменных смол — не более 70, а диатомитовых носителей для газожидкостной хроматографии — менее 10 м2/г. П. у. характеризует дисперсность порошкообразных материалов: минеральных вяжущих веществ, наполнителей, пигментов, пылевидного топлива и др. Величина их П. у. обычно находится в пределах от десятых долей до нескольких десятков м2/г. П. у. чаще всего определяют по количеству адсорбированного материалом инертного газа и по воздухопроницаемости слоя порошка или пористого материала. Адсорбционные методы позволяют получать наиболее достоверные данные.
Лит.: Грег С., Синг К., Адсорбция, удельная поверхность, пористость, пер. с англ., М., 1970; Коузов П. А., Основы анализа дисперсного состава промышленных пылей и измельченных материалов, 2 изд., Л., 1974.
Л. А. Шиц.
Разделения методы в аналитической химии, совокупность операций, применяемых с целью обнаружения и количественного определения какого-либо элемента (вещества) в сложном по составу анализируемом материале. Р. м. необходимы, поскольку большинство аналитических методов недостаточно избирательны. При разделении ионов элементов используют групповые реагенты, позволяющие упростить трудноразрешимую задачу анализа сложных смесей. Для разделения применяют осаждение (см. Осаждения способ), экстракцию, хроматографию, дистилляцию, а также др. способы.
Сефадексы, синтетические производные полисахарида декстрана, в полимерные молекулы которых введены поперечные «сшивки», образующие трёхмерную сетку с порами заданного размера (т. н. молекулярные сита). С. производят в виде гранулированного порошка с размером частиц 20—300 мкм и классифицируют в зависимости от величины пор. При набухании в воде или солевых растворах С. образуют гели, представляющие двухфазную систему растворителя внутри гранул и в свободном объёме. При хроматографии на колонках из С. молекулы большего размера фильтруются быстрее, не задерживаясь в порах набухшего геля, а меньшего — «застревают» в порах и выходят позже. На этом основано широкое применение С. в химии и биохимии для разделения (фракционирования) смесей веществ с молярной массой 102—2×105, определения молярных масс глобулярных белков и ферментов, для обессоливания высокомолекулярных соединений и их концентрирования. Включением в С. ионообменных групп получают иониты с высокой разделяющей способностью, используемые для очистки биополимеров. С. применяются в промышленности при производстве витаминов, антибиотиков и др.
Н. Н. Чернов.
Силикагель, высушенный гель поликремниевой кислоты, твёрдый гидрофильный сорбент. По химическому составу С, — двуокись кремния SiO2 (кремнезём), по структуре — высокопористое тело, образованное мельчайшими сросшимися сферическими частицами. Получают С. следующим образом: действуют на раствор силиката натрия или калия (жидкое стекло) соляной или серной кислотой, а затем затвердевший продукт разламывают на куски, промывают водой, сушат, измельчают, фракционируют и прокаливают для полного удаления влаги. Товарный С. выпускают в виде зёрен или шаровидных гранул размером от 5—7 до 10-2 мм. Различные марки С. имеют средний эффективный диаметр пор 20—150 Å и поверхность удельную 102—103 м2/г. С. используют для поглощения паров воды и органических растворителей, адсорбционной очистки неполярных жидкостей, в газовой и жидкостной хроматографии для разделения спиртов, аминокислот, витаминов, антибиотиков и др. Крупнопористые С. применяются как носители катализаторов.
Лит.: Кольцов С. И., Алесковский В. Б., Силикагель, его строение и химические свойства, Л., 1963.